
Is machine learning taking over drug discovery?
Not yet.

Five years out from the launch of AlphaFold, we’ve heard precious little about the transformative impact that AI was promised to have on drug discovery. And if we look at the numbers for AI-designed drugs that have entered the market (0), they don’t look quite promising.
Now, everyone knows that Big Pharma is a lumbering behemoth infested with regulatory burden, so plausibly, the technology is very promising and useful but hasn’t yet made it through modern-day drug pipelines. If this were true, we’d expect a massive influx of AI-generated drugs once the first wave makes it through the pipeline and we break down the major sources of regulatory impediments. The number of drugs approved by the FDA has hovered around 50/year for the past decade, but we might imagine a ChatGPT moment in drug discovery that sends us on an exponential curve to Solving All Disease.

Alternatively, it’s possible that the models are helpful but not yet useful enough to automate all drug discovery, in which case we might see steadily more AI-generated drugs hit the market, with no inflection point or guaranteed path to automation.
The first story is the one that many drug discovery companies are selling.
The true story is closer to the second scenario and a combination of other factors. I’d argue we shouldn’t anticipate a massive step function in the number of drugs produced, at least not within the next decade:
I. AI drug discovery companies are largely focused on automating preclinical drug development which makes up roughly 1/3 of the cost and time of drug development, however, existing models still require a significant degree of target-specific tuning. A significant impediment to the accuracy of these models is a deficiency of experimental data, especially from human trials, so improving the efficiency of clinical trials must happen in parallel with and enable advancements in the ML stack.
II. The remaining 2/3 of drug development—clinical trials—can only be accelerated marginally through the introduction of better technological stacks. Most of the slowdown comes from regulatory policy and the necessity of long-term trials.
To be clear, should certainly expect the number of AI-generated drugs entering clinical trials to increase—the number has been roughly doubling each year since 2021 alongside a 30% increase in the number of partnerships between traditional pharma and AI tech firms! However, the emphasis here is that we need more innovative solutions at the clinical trial stage. Trials take longer to start, enroll slower, and cost more today than they did 3-5 years ago.
Let’s start off by getting a deeper understanding of the drug discovery process.
How does drug discovery work?
Drugs currently take 10-15 years on average to go from discovery to deployment.
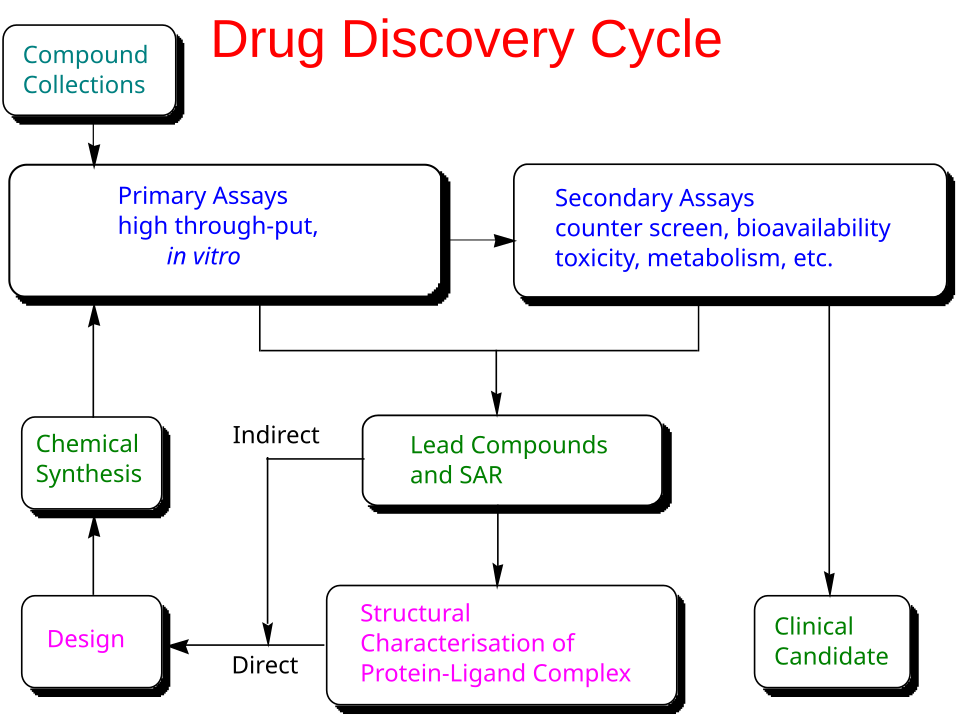
Most therapeutics work by modulating a biological target—commonly a human protein, a pathogen protein, or nucleic acids—though some medicines act primarily through broader physicochemical or reactive chemical mechanisms. The first step of drug discovery is, therefore, target discovery. Targets can be proposed from large-scale observational data (for example, multiomics and clinical datasets that implicate pathways in disease) and strengthened through causal evidence, such as genome-wide association studies in humans or functional perturbation screens using CRISPR-based interference. Many targets already have partial characterization in the literature and public resources1, including Pharos and the Human Protein Atlas, but poorly characterized targets are typically higher risk because they lack reliable assays, reagents, and safety context. In practice, target discovery often involves integrating a massive amount of fragmented evidence across papers, databases, and other unstructured sources.
Once a target is prioritized for a program, preclinical discovery and development lasts 4-5 years on average. The process can be summarized as an iterative loop of drug design, synthesis, and assays until the right combination of properties are achieved. The properties to optimize include the drug’s intended biological activity (potency, selectivity, and evidence of target engagement), its behavior in the body (absorption, distribution, breakdown, and clearance), its safety margin, and its developability (stability, formulation, and manufacturability), with somewhat different priorities for small molecules and biologics.
After a satisfactory candidate has been identified, the developers must then undergo the Investigational New Drug (IND) application before proceeding to clinical trials.
The clinical component of drug discovery typically lasts 6-10 years and consists of 4 phases.
The process has a success rate of less than 10%2, with 40% and 50% of failures due to a lack of clinical efficacy (the drug wasn’t able to produce its intended effect in people, 30% due to unmanageable toxicity or side effects, 10%-15% due to poor pharmacokinetic properties (how well a drug is absorbed by and excreted from the body), and 10% due to lack of commercial interest and poor strategic planning.
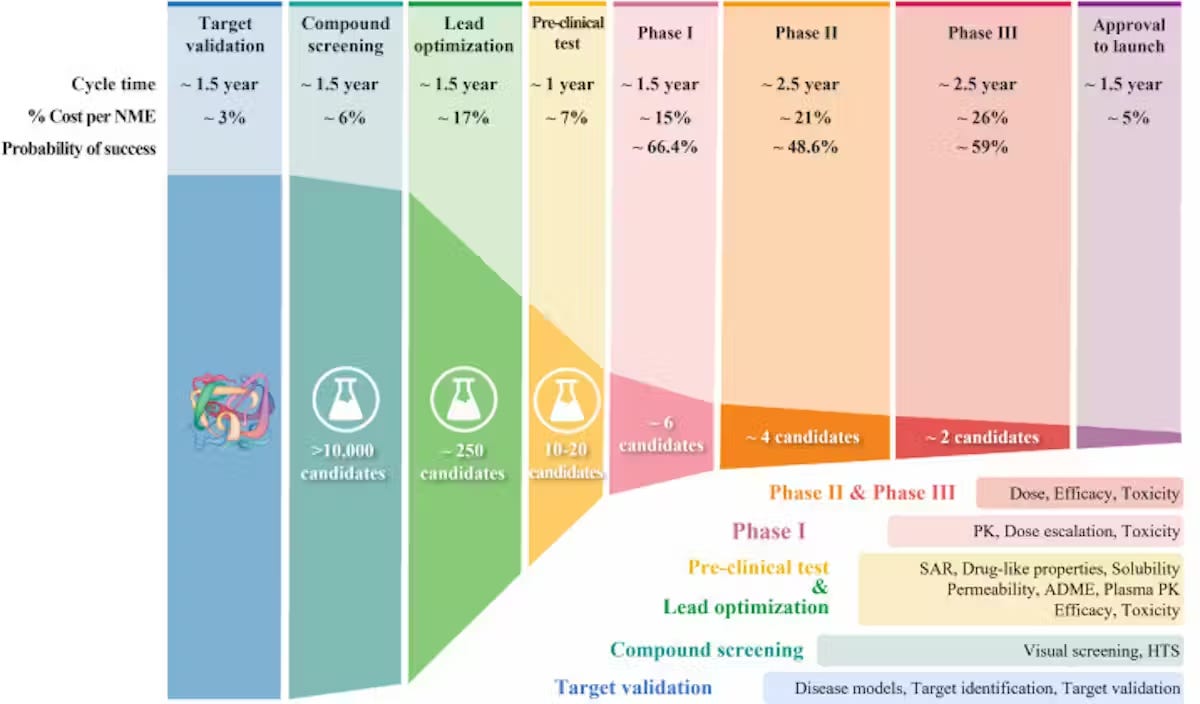
This leads us to the main metrics of success for assessing the efficiency of drug discovery pipelines:
Validated hit rate per target (and how many designs screened to get a hit)
Cycle time: how long it takes for a drug candidate to become a synthesized compound and be validated for activity
Lead optimization efficiency: potency/selectivity gains per design–make–test round
Clinical progression: % success rate of passing each clinical stage
What is the current state of ML in drug discovery?
It’s important to recognize here that the use of ML in drug discovery can encompass both 1) AlphaFold and adjacent protein prediction systems and 2) LLMs. Applying machine learning to biological data is a research direction that’s been around since before GPT-3 came out in 2020. While people are now racing to train transformers on all types of biological data, we cannot directly apply the same scaling laws to biology as to text, and the most successful ML models in the field have required the incorporation of bespoke biological insights, such as the explicit encoding of evolutionary signal into AlphaFold2. I’d venture to say that ML in biology as a field has largely progressed at its own pace and will continue to do so in the upcoming years in tandem with innovations in high throughput assaying and wet-lab automation.
Impact of LLMs
LLMs have already proven to be useful in large-scale analysis of unstructured data. The computational component of target discovery is largely shifting from slow, manual synthesis of evidence from papers and patient datasets to LLM-based methods to prioritize targets and propose mechanistic hypotheses.
Moreover, platforms like Deep 6 AI market accelerate the process of matching patients to clinical trials from EMR data.
And as is the case across fields, individuals who are able to leverage AI tools in their work can experience massive productivity gains. The illegible minute enhancements that LLMs can enable for knowledge workers cannot be understated.
Impact of Protein Models
Protein machine-learning models used in drug discovery mostly fall into two buckets:
Structure models: predict the 3D arrangement of atoms for a single protein or a multi-molecule complex (for example, a protein bound to DNA or a small-molecule ligand).
Property models: predict experimentally relevant behaviors directly from sequence or structure—such as binding strength, stability, solubility, clearance, toxicity risk, and other absorption, distribution, metabolism, and excretion properties.
After a biological target is identified, a common next step is screening: testing (computationally and experimentally) large libraries of candidate small molecules to find an initial “hit” that binds the target. Historically, a major bottleneck has been obtaining high-quality 3D structures of the target, since experimental techniques like X-ray crystallography and nuclear magnetic resonance can be slow and expensive, with costs upwards of $100,000
Modern structure prediction models reduce this bottleneck by producing useful structural hypotheses quickly. AlphaFold 3, for example, predicts structures for assemblies that can include proteins, nucleic acids, and small molecules. Related efforts include RoseTTAFold All-Atom, open-source Boltz-style models, Pearl, and Chai-1.
This process is followed by docking and pose prediction, a process of narrowing down the candidates through the strength of their relationship with the target molecule. DiffDock reframes docking as a generative (diffusion) process that samples plausible ligand poses conditioned on the target structure.
Each drug design then undergoes experimental testing and optimization. The initial molecule can be tweaked hundreds of thousands of times to find a candidate with improved potency, stability, pharmacokinetic properties, and safety, while still being feasible to synthesize. Property prediction models are important during this phase to provide fast, approximate feedback on multiple objectives.
Genesis has taken the approach of designing a full closed-loop platform that orchestrates proposing new molecules, property prediction, and docking and molecular dynamics. Their ensemble of models includes Pearl (3D structure generation), an model for ADME property prediction, and physical simulations for potency and selectivity estimation.
So where are all the AI drugs?
In clinical trials! A June 2024 analysis estimated 67 AI-discovered or AI-enabled drugs in clinical trials at the end of 2023, with 45 in Phase I (several months), 19 in Phase II (several months to two years), and 2 in Phase III (1-4 years).
This is out of around nearly 2,000 drugs entering clinical trials total.
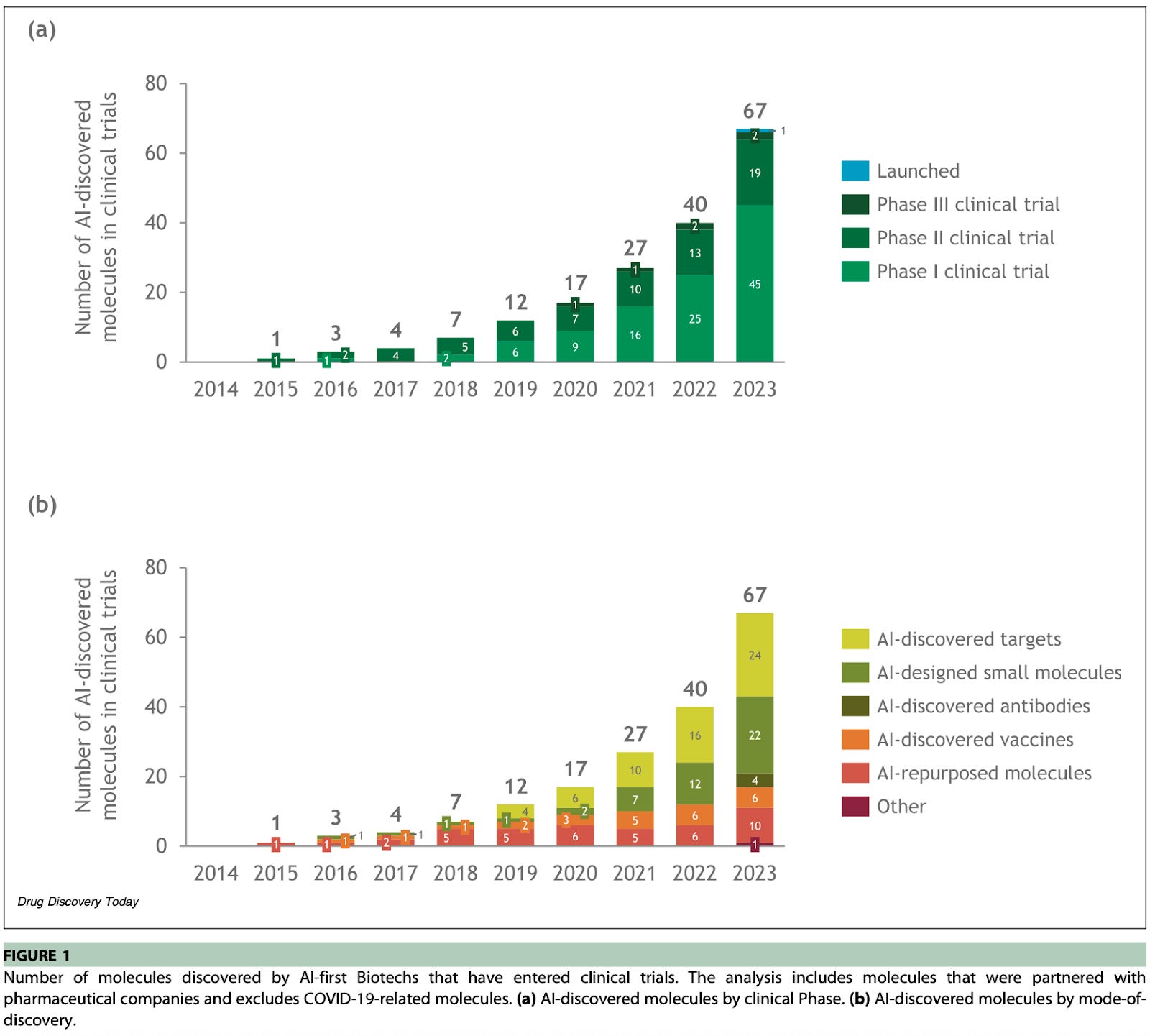
The subsequent FDA review takes on the order of 6-10 months, which means we might not expect the first AI-designed drugs to hit the market until 2026 or early 2027.
A full list of AI-generated drugs in clinical trials is provided in the appendix, and they originate from companies like Exscientia, Recursion, Benevolent AI, Insilico, and more. Notably, Exscientia EXS4318 was designed in ~11 months (a significant improvement from the industry 4-5 year average!), and the hit was the 150th novel compound synthesized in that program. The drug was described to be a particularly difficult kinase target because it requires high potency and very high selectivity, and should have an sustained effect at a low daily human dose.
Finally, Isomorphic Labs, the DeepMind spinout that inherited AlphaFold, has started setting up clinical infrastructure in Boston and will have drug candidates starting human trials in early 2026!
Will ML solve Eroom’s Law?
The number of new drugs approved per billion dollars spent has been halving every 9 years for the past 70 years.
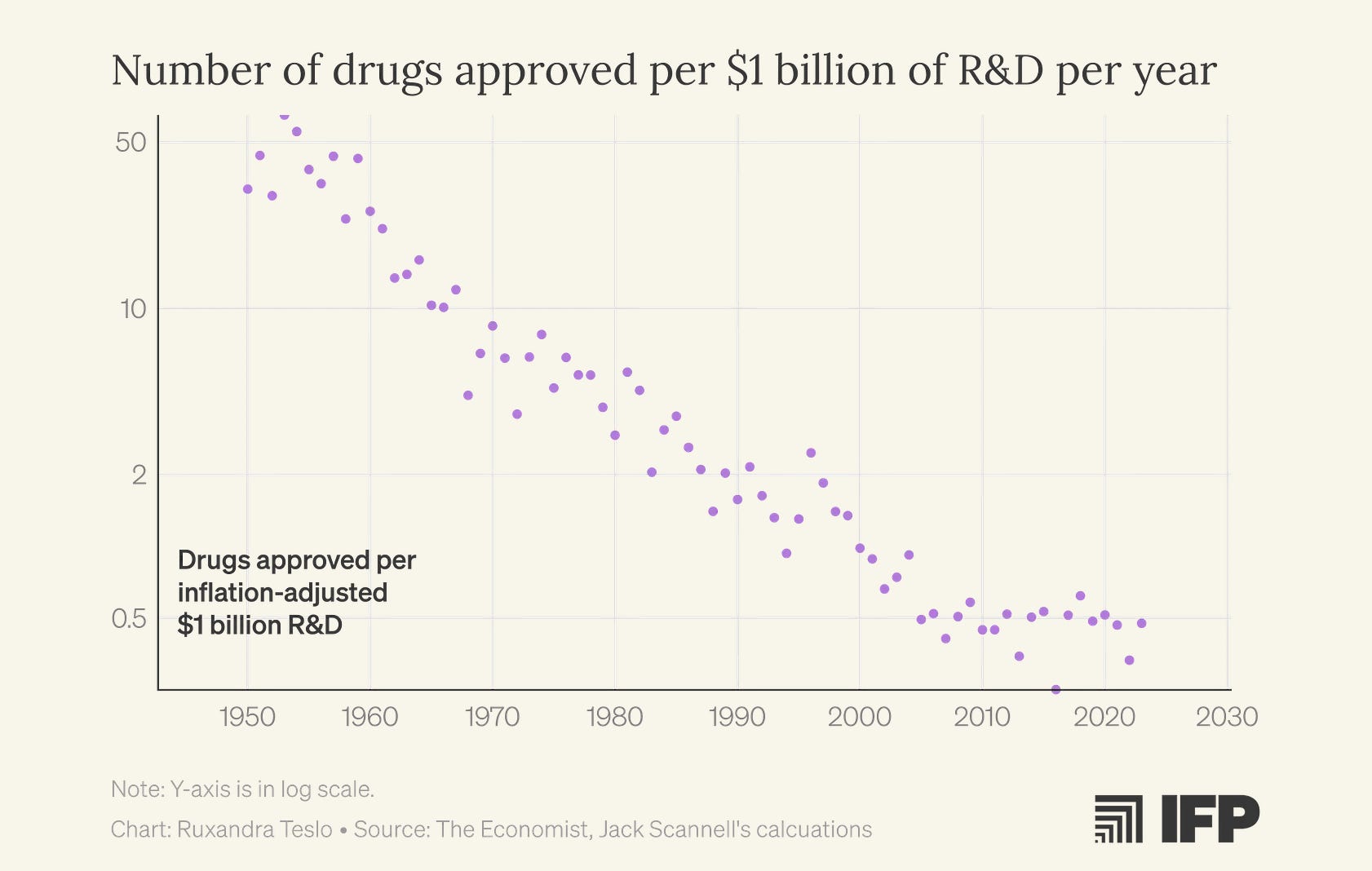
Ruxandra discusses in detail how this phenomenon, referred to as Eroom’s Law, might be directly tied to the increasing popularity of ML approaches. Even when the underlying science wasn’t yet mature enough to support robust, predictive mechanistic models, ML promised to solve molecular dynamics and drug design. The focus on computational methods detracted from iterative, empirical exploration.
From a financial perspective, I find this story somewhat difficult to believe. The amount of attention and money invested into AI-first drug discovery approaches remains a tiny fraction of the investment poured into the industry as a whole. Depending on whether you measure “AI drug discovery” as startup funding or tool market spend, you’re only looking at roughly ~1% to ~2.5% of the industry’s annual R&D magnitude.
That said, it’s certainly true that venture-backed industries love clean technological narratives, especially ones that suggest a single “core scientific breakthrough.” Teams are often incentivized to over-index on building more novel solutions and more advanced models, and under-index on the underlying bedrock of measurement, assay design, and clinical execution.
Biotechnology as a field doesn’t move like software. It’s not a field in which two college students can entirely break the current paradigm. Such a phenomenon was almost uniquely suited to the internet age because of the nature of the technology. Software distribution is nearly free, iteration is instantaneous, and network effects make progress self-accreting. Drug discovery and validation, on the other hand, must happen in the real world and face the frictions of wet lab, human trials, and regulations.
And biology is complex and filled with esoteric edge cases that break clean abstractions. Even researchers at the frontier of protein and molecular modeling still point to data, not architectures, as a core bottleneck. Building a predictive “map” that captures the specificity of our historically hand-curated mechanisms remains an open challenge.
Still, I’m optimistic. The last decade’s suboptimalities have pushed the field toward the right lessons. We’re seeing more serious partnerships with Big Pharma, more in-house wet-lab capacity, and a growing respect for the unglamorous work that actually closes the loop: better assays, better datasets, and better clinical infrastructure. AlphaFold-class models are extraordinary, but structure and sequence models only plausibly automate up to 30% of the drug discovery process. The rest will be won by less sexy, collective efforts to increase the throughput and quality of real-world testing.
It’s estimated that we have sufficient chemical data on about 69% of the known human proteome.
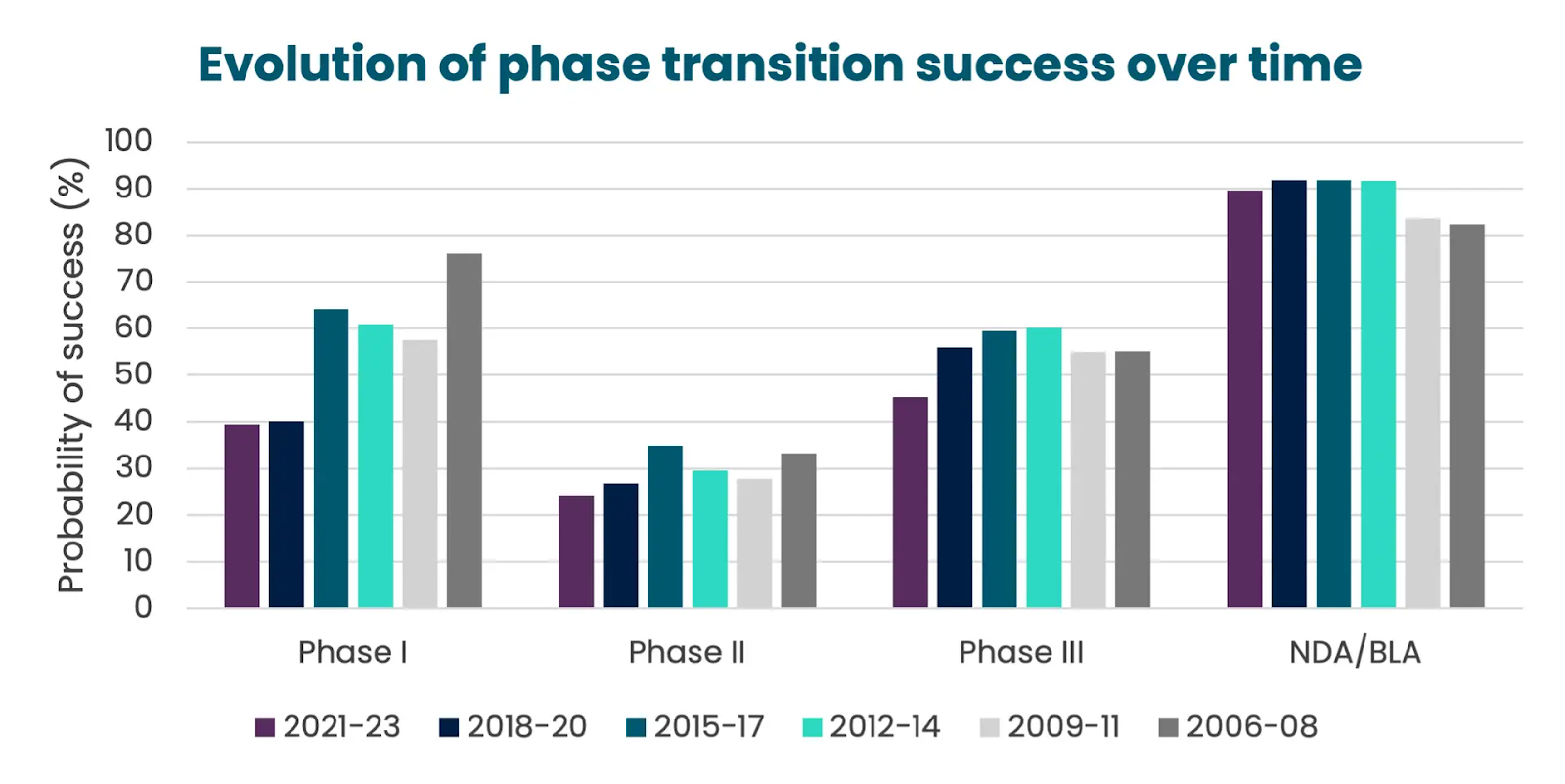
In 2023, the rates were success for drugs navigating across the different clinical phases looked like the following:
Phase I → Phase II: ~47%
Phase II → Phase III: ~28%
Phase III → Approval: ~55%
Filing → Approval: ~92%